
- About ACNS
- Meetings
- Annual Meeting and Courses
- Principles of Clinical Neurophysiology Courses
- Claim CME: Past Meetings
- Meetings & Programs Calendar
- Endorsed Courses & Symposia
- Education
- Practice
- Research
- Advocacy
- Membership
Featured Case
Contributed by Puneet Jain, MD, DM
The Hospital for Sick Children, Toronto, Canada
This boy was born to a non-consanguineous couple at 40 weeks gestation with an unremarkable antenatal history and no evidence of birth asphyxia. However, from the first day of life he demonstrated persistent lethargy and poor feeding. Examination showed normal head-circumference, encephalopathy with poor state variability, global hypotonia and depressed deep tendon reflexes. On the sixth day of life he was noted to have very frequent brief body jerks involving the distal extremities during both wakefulness and sleep. He also experienced frequent hiccups and occasional brief episodes of tonic posturing of the left arm. Continuous EEG monitoring was initiated to characterize these events.
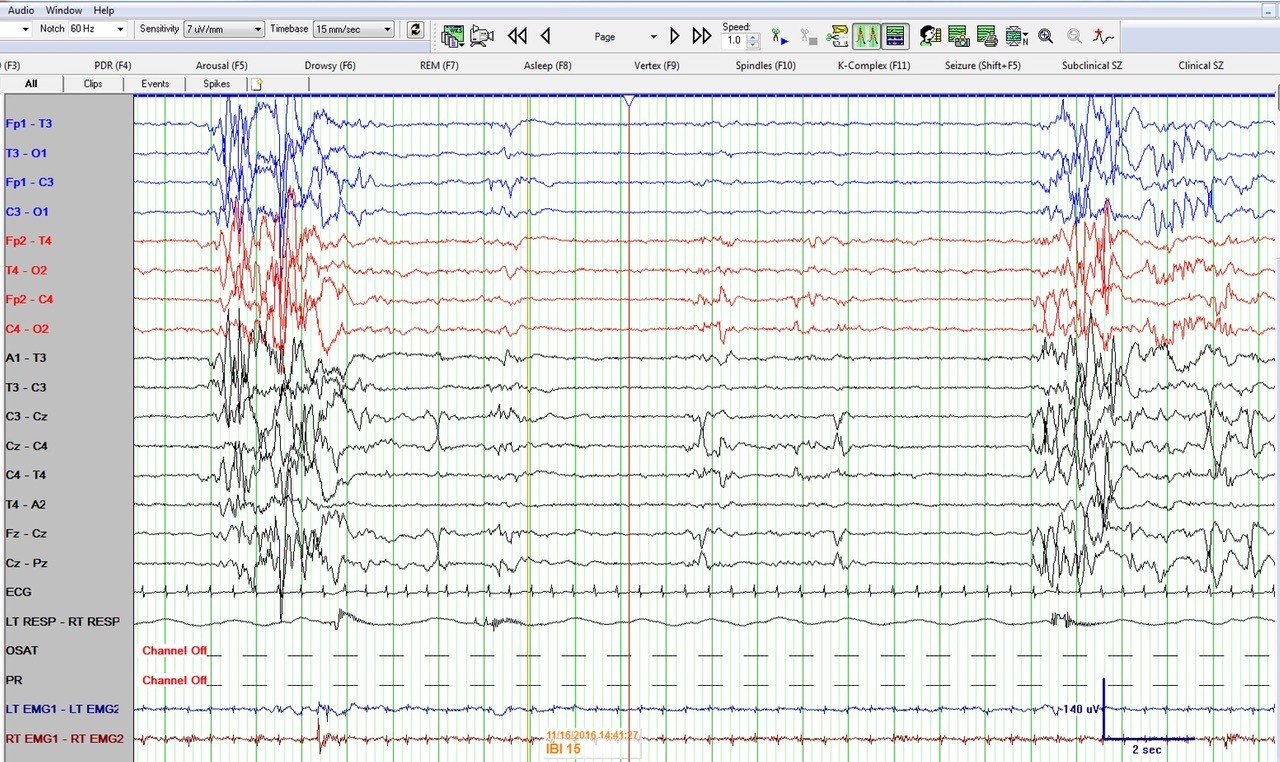
Figure 1: Neonatal bipolar montage with a time base 15 mm/sec, sensitivity 7 µV/mm, high frequency filter at 70 Hz, low frequency filter at 1 Hz and sampling rate of 256 Hz
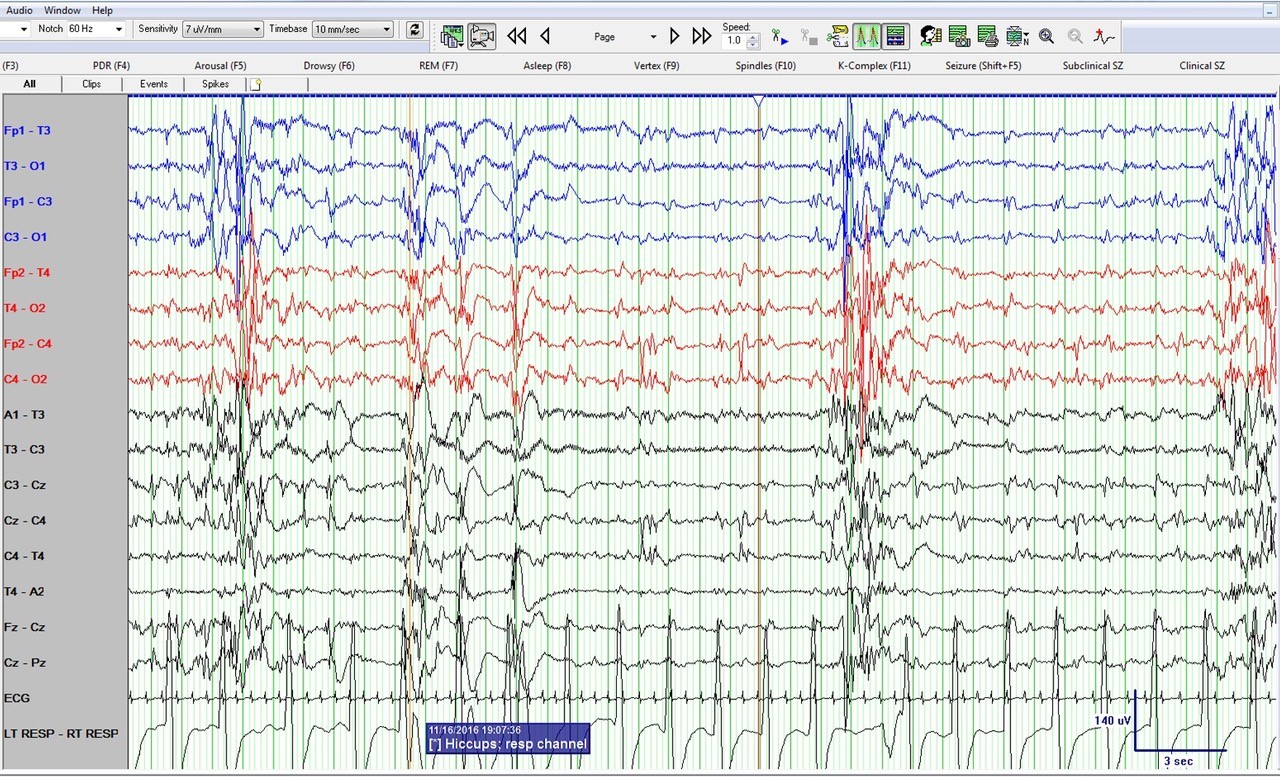
Figure 2: Neonatal bipolar montage with a time base 10 mm/sec, sensitivity 7 µV/mm, high frequency filter at 70 Hz, low frequency filter at 1 Hz and sampling rate of 256 Hz
Question: What do these EEG epochs represent?
Answer & Discussion
Figure 1 shows a burst suppression pattern with inter-burst interval of around 15 seconds and amplitude < 10 µV. Also note sharp waves at Cz (negative) and C4 (positive) during the suppression phase. Figure 2 again shows the burst suppression pattern, but also note that the baby was having rhythmic ~1 Hz hiccups during this epoch as evident on the respiratory channel (and confirmed by video) but with no accompanying EEG change.
In view of the boy’s presentation of neonatal encephalopathy, fragmentary multifocal myoclonia, frequent hiccups and burst suppression pattern on EEG, the possibility of glycine encephalopathy (a.k.a. non-ketotic hyperglycinemia) was considered.
Laboratory investigations showed normal blood sugar, calcium, pH, lactate, ammonia and urine organic acids. CSF glyine [321 µmol/L (normal range, 6-19)] and plasma glycine [1571 µmol/L (normal range, 178-248)] were markedly elevated. The CSF/Plasma glycine ratio was also elevated at 0.2:1 (>0.08 is highly suggestive of glycine encephalopathy). Urinary glycine was also elevated [13242 mmol/mol Cr (normal range, 563-2563)]. MRI brain showed bilateral hyperintensities on T2-weighted sequences throughout the cerebral white matter, posterior limbs of the internal capsule, subthalamic region, mid-brain and within the dorsal tegmental tracts.
These investigations confirmed the diagnosis of glycine encephalopathy with early myoclonic encephalopathy (EME). The baby was treated with multiple anti-seizure drugs, sodium benzoate, carnitine, dextromethorphan and supportive care.
Glycine encephalopathy is an autosomal-recessive disorder caused by deficient activity of the multimeric glycine-cleavage enzyme with resultant accumulation of excessive of glycine in all body tissues including the brain. Most patients present in the neonatal period (usually within in the first hours to days of life) with progressive lethargy, hypotonia, and myoclonic jerks leading to apnea and often death. The diagnosis is made by elevated CSF-to-plasma glycine ratio, liver enzymatic estimation or genetic testing. There is no effective treatment.
EME is a rare epileptic-encephalopathy. Seizures usually begin in the first month of life. Erratic, fragmentary myoclonus is the defining seizure type. Myoclonus often affects the face and limbs, and is usually very frequent. Antenatal myoclonus has also been reported. Other seizure semiologies may include focal seizures (eye deviation, apnea, clonic, asymmetric tonic) and rarely tonic spasms. Seizures remain refractory to treatment with anti-seizure medications. There is severe psychomotor retardation. EME may evolve into West syndrome or epilepsy with multiple-independent-spike-foci (MISF). More than half of patients with EME die before age 2 years. Many cases of EME are familial. Metabolic causes predominate, although the etiology remains unknown in the majority of cases. Reported causes include glycine encephalopathy, organic acidemias, sulphite and xanthine oxidase deficiency, molybdenum co-factor deficiency, Menkes disease and Zellweger syndrome.
The inter-ictal EEG in patients with EME characteristically demonstrates a burst-suppression pattern, which consists of bursts lasting 1 to 5 seconds alternating with periods of suppression lasting for 3 to 10 seconds. This pattern is often more prominent during sleep, or may occur exclusively during sleep. Compared to Ohtahara syndrome, patients with EME typically have longer inter-burst intervals, asynchronous bursts and more multifocal spikes during the suppression phase. The EEG during wakefulness usually shows slow background activity with frequent multifocal spike-waves.
Myoclonias are typically not associated with ictal EEG changes, but may coincidentally coincide with bursts of the burst-suppression pattern. Focal seizures may have variable ictal EEG patterns including rhythmic alpha or theta activity, or irregular spike-wave activity. Tonic spasms are accompanied by generalized voltage attenuation.
Further reading