
- About ACNS
- Meetings
- Education
- Practice
- Research
- Advocacy
- Membership
Contributed by: Hicham Dabaja, MD; Bernice Casella, APRN, CNP, Lily Wong-Kisiel, MD
A 5-year-old boy with a history of febrile seizures presented for a routine EEG as part of ongoing epilepsy follow-up. He had experienced three febrile seizures at 17, 24, and 31 months of age. The initial seizure was classified as complex febrile, characterized by two seizures within a 24-hour period, while the subsequent episodes were simple febrile seizures. Later, the patient developed afebrile generalized tonic-clonic seizures, with three episodes lasting 1–2 minutes each. These occurred in the context of significant sleep disruption following prolonged transpacific travel. At that time, a short-term awake and asleep EEG revealed no abnormalities. He was started on levetiracetam, achieving seizure freedom thereafter. During the current EEG, while performing hyperventilation, the patient exhibited generalized semirhythmic 2–3 Hz high-amplitude delta activity after approximately three minutes. This pattern was associated with behavioral arrest, as the patient ceased hyperventilating. No accompanying myoclonus or other adventitial movements were observed. The episode resolved spontaneously, with the patient returning to baseline immediately afterward. When prompted by the technologist one second before the resolution of the generalized delta with the question, "Are you really, really dizzy?" the patient responded appropriately with "yes."
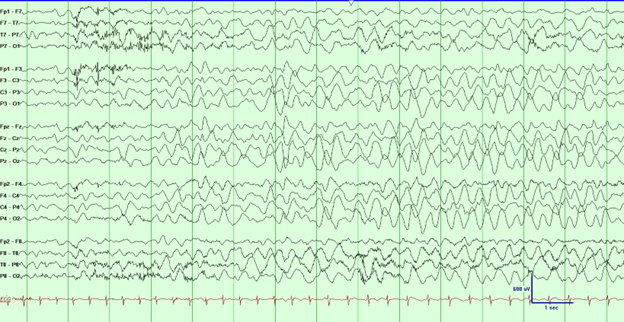
Figure 1: EEG tracing associated with behavioral arrest. EEG filter settings: LFF: 1 Hz, HFF: 70 Hz, Notch: Off, Sensitivity: 30 μV/mm, Timebase: 30 mm/sec.
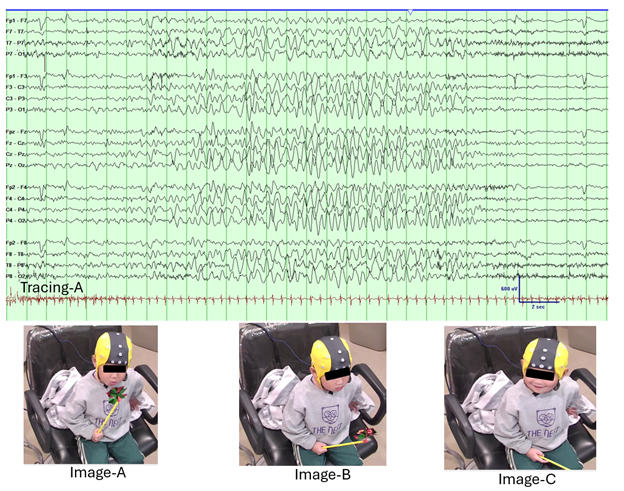
Figure 2: This figure illustrates the EEG and clinical correlation during hyperventilation-induced high-amplitude rhythmic slowing (HIHARS). In EEG Tracing A, generalized semirhythmic 2–3 Hz high-amplitude delta activity is observed during hyperventilation (EEG filter settings: LFF: 1 Hz, HFF: 70 Hz, Notch: Off, Sensitivity: 30 μV/mm, Timebase: 15 mm/sec), consistent with HIHARS. Image A shows the patient actively blowing on a pinwheel, initiating hyperventilation during the EEG recording. In Image B, the patient has stopped blowing on the pinwheel and displays behavioral slowing, paralleling the delta activity seen on the EEG. Finally, Image C captures the patient returning to baseline, smiling and engaging with the technologist after verbally responding to the question, “Are you really really dizzy?”
Question 1: What is the characteristic EEG pattern associated with hyperventilation-induced high-amplitude rhythmic slowing (HIHARS)?
Question 2: Which clinical feature helps distinguish HIHARS from absence seizures during hyperventilation?